…Before a Droplet Hits the Water was inspired by an image that captured the moment of what the title depicts. A paradox is created by the frozen frame of this ordinary event. Although the droplet may be the focus of attention due to its dynamic nature, it is the water that holds the tension yet to be broken. It is a strange moment when the perception and the expectation evenly take up the brain. The music is a reflection of this scenario in the aural dimension. The foreground is constantly but slowly modulated while the background undergoes a stealthy shift, leaving the audience in an uncertain space between establishing their expectation and it being already satisfied.
Author: Xavier
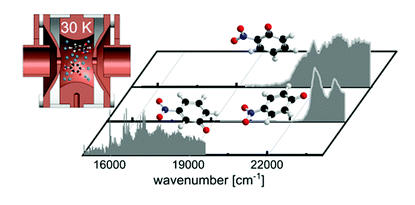
Intrinsic photophysics of nitrophenolate ions studied by cryogenic ion spectroscopy
The intrinsic photophysics of nitrophenolate isomers (meta, para, and ortho) was studied at low temperature using photodissociation mass spectrometry in a cryogenic ion trap instrument. Each isomer has distinct photophysics that affects the excited state lifetimes, as observed experimentally in their spectroscopic linewidths. Visible-light-induced excitation of m-nitrophenolate gives rise to well-resolved vibronic features in the spectrum of the S1 state. The para and ortho isomers have broad spectra – even at cryogenic temperatures – due to their shorter excited state lifetimes and spectral congestion. We present computational evidence for mixing of the first and second excited states of o-nitrophenolate, leading to significant additional broadening in the experimental spectrum.
DOI: 10.1039/C8CP06078A
Leah G. Dodson,a Wyatt Zagorec-Marks,b Shuang Xu,c James E. T. Smithb and J. Mathias Weberb
aJILA and NIST, University of Colorado, 0440 UCB, Boulder, USA
bJILA and Department of Chemistry, University of Colorado, 0440 UCB, Boulder, USA
cJILA and Department of Physics, University of Colorado, 0440 UCB, Boulder, USA
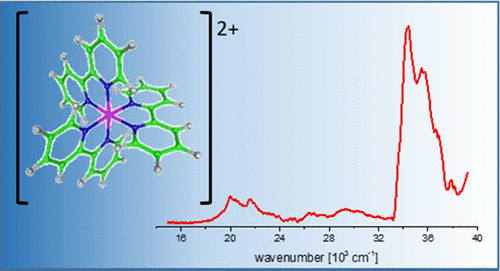
Electronic Spectra of Tris (2, 2′-bipyridine)-M (II) Complex Ions in vacuo (M= Fe and Os)
We measured the electronic spectra of mass-selected [M(bpy)3]2+ (M = Fe and Os, bpy = 2,2′-bipyridine) ions in vacuo by photodissociation spectroscopy of their N2 adducts, [M(bpy)3]2+·N2. Extensive band systems in the visible (predominantly charge transfer) and near-ultraviolet (ππ*) spectral regions are reported. The [M(bpy)3]2+·N2 target ions were prepared by condensing N2 onto electrosprayed ions in a cryogenic ion trap at ca. 25 K and then mass-selected by time-of-flight mass spectrometry. The electronic photodissociation spectra of the cold, gas-phase ions closely reflect their intrinsic properties, i.e., without perturbation by solvent effects. The spectra are interpreted using time-dependent density functional theory calculations both with and without accounting for relativistic effects.
DOI: 10.1021/acs.inorgchem.7b00620
Shuang Xu†, James E. T. Smith‡, and J. Mathias Weber‡
† JILA and Department of Physics, University of Colorado, Boulder, Colorado 80309-0440, United States
‡ JILA and Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309-0440, United States
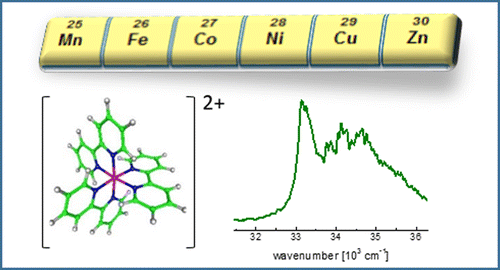
UV Spectra of Tris (2, 2′-bipyridine)–M (II) Complex Ions in vacuo (M= Mn, Fe, Co, Ni, Cu, Zn)
We present electronic spectra in the π–π* region of a series of tris(bpy)–M(II) complex ions (bpy = 2,2′-bipyridine; M = Mn, Fe, Co, Ni, Cu, Zn) in vacuo for the first time. By applying photodissociation spectroscopy to cryogenically cooled and mass selected [MII(bpy)3]2+ ions, we obtain the intrinsic spectra of these ions at low temperature without perturbation by solvent interaction or crystal lattice shifts. This allows spectroscopic analysis of these complex ions in greater detail than possible in the condensed phase. We interpret our experimental data by comparison with time-dependent density functional theory.
DOI: 10.1021/acs.inorgchem.6b02054
Shuang Xu†, James E. T. Smith‡, and J. Mathias Weber‡
† JILA and Department of Physics, University of Colorado, Boulder, Colorado 80309-0440, United States
‡ JILA and Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309-0440, United States

Hydration of a Binding Site with Restricted Solvent Access: Solvatochromic Shift of the Electronic Spectrum of a Ruthenium Polypyridine Complex, One Molecule at a Time
We report the electronic spectra of mass selected [(bpy)(tpy)Ru–OH2]2+·(H2O)n clusters (bpy = 2,2′-bipyridine, tpy =2,2′:6′2″-terpyridine, n = 0–4) in the spectral region of their metal-to-ligand charge transfer bands. The spectra of the mono- and dihydrate clusters exhibit partially resolved individual electronic transitions. The water network forming at the aqua ligand leads to a rapid solvatochromic shift of the peak of the band envelope: addition of only four solvent water molecules can recover 78% of the solvatochromic shift in bulk solution. The sequential shift of the band shows a clear change in behavior with the closing of the first hydration shell. We compare our experimental data to density function theory (DFT) calculations for the ground and excited states.
Shuang Xu†, James E. T. Smith‡, and J. Mathias Weber‡
† JILA and Department of Physics, University of Colorado, Boulder, Colorado 80309-0440, United States
‡ JILA and Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309-0440, United States
The electronic spectrum of cryogenic ruthenium-tris-bipyridine dications in vacuo
We report the electronic spectrum of the prototypical ruthenium coordination complex Ru(bpy)3 2+ (bpy = 2, 2′-bipyridine) by messenger tagging with N2 in a cryogenic ion trap and photodissociation spectroscopy of mass selected Ru(bpy)3 2+ ⋅ N2 ions. We observe individual electronic bands and groups of bands with unprecedented detail, particularly in the usually unresolved metal-to-ligand charge transfer region of thespectrum. By comparing our experimental results with time-dependent density functional theory, both with and without spin-orbit interaction [Heully et al., J. Chem. Phys. 131, 184308 (2009)], we are able to assign the spectrum of the isolated ion.
DOI: 10.1063/1.4955262
Shuang Xu†, James E. T. Smith‡, and J. Mathias Weber‡
† JILA and Department of Physics, University of Colorado, Boulder, Colorado 80309-0440, United States
‡ JILA and Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309-0440, United States
Cat, op.23
A concerto
I realized that I was often in a hurry to finish a composition because I was afraid I would lose my consistent thoughts if it took too long. However, historically, it’s rarely the case. Masterworks often took years to finish even by the maestri. In 2011 I wanted to take a different approach. I would take my time to compose and to polish and write a real delicate piece.
The choice of subject and genre didn’t take long though, since I decided to dedicated it to my “feline” girlfriend, who is a violinist. Being a cat lover myself and inspired all cat videos online, I came up with this idea of employing real cats into this piece. But that’s more or less a gimmick. The actual music is supposed to depict the life of a cat.
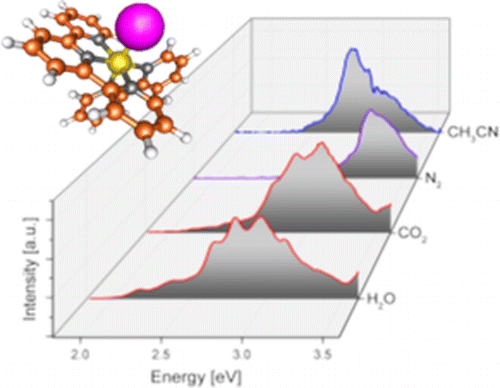
Ligand Influence on the Electronic Spectra of Dicationic Ruthenium-Bipyridine-Terpyridine Complexes
We report electronic spectra of a series of ruthenium polypyridine complexes of the form [(trpy)(bipy)RuII–L]2+ (bipy = 2,2′-bipyridine and trpy = 2,2′:6′,2″-terpyridine), where L represents a small molecular ligand that occupies the last coordination site. Species with L = H2O, CO2, CH3CN, and N2 were investigated in vacuo using photodissociation spectroscopy. All species exhibit bright metal-to-ligand charger transfer (MLCT) bands in the visible and near UV, but with different spectral envelopes and peak energies, encoding the influence of the ligand L on the electronic structure of the complex. Several individual electronic bands can be resolved for L = H2O and CO2, while the spectra for L = N2 and CH3CN are more congested, even at low ion temperatures. The experimental results are discussed in the framework of time-dependent density functional theory.
Shuang Xu†, James E. T. Smith‡, and J. Mathias Weber‡
† JILA and Department of Physics, University of Colorado, Boulder, Colorado 80309-0440, United States
‡ JILA and Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309-0440, United States
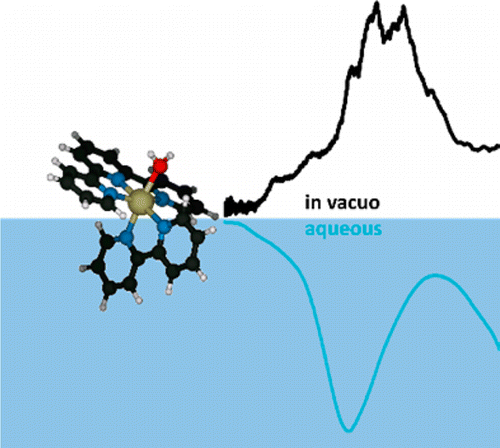
Absorption Spectrum of a Ru(II)-Aquo Complex in Vacuo: Resolving Individual Charge-Transfer Transitions
† JILA, University of Colorado, 440 UCB, Boulder, Colorado 80309, United States
‡ Department of Physics, University of Colorado, Boulder, Colorado 80309, United States
§ Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, United States
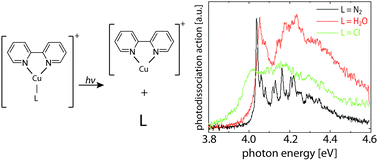
Ligand influence on the electronic spectra of monocationic copper–bipyridine complexes
We present photodissociation spectroscopy and computational analysis of three monocationic Cu–bipyridine complexes with one additional ligand of different interaction strength (N2, H2O and Cl) in the visible and UV. All three complexes show similar ππ* bands with origins slightly above 4 eV and vibrational band contours that are due to bipyridine ring deformation modes. Experiments at low temperature show that excited-state lifetime is the limiting factor for the width of the vibrational features. In the case of Cl as a ligand, there is a lower lying bright ligand-to-ligand charge-transfer state around 2.75 eV. The assignment of the transitions was made based on equation-of-motion coupled-cluster calculations. While the nature of the ligand does not significantly change the position of the bright ππ* state, it drastically changes the excited-state dynamics.
DOI: 10.1039/C5CP05063D
aJILA and Department of Physics, University of Colorado, Boulder, USA
bDepartment of Chemistry, University of Southern California, Los Angeles, USA
cJILA and Department of Chemistry and Biochemistry, University of Colorado, Boulder, USA
* weberjm@jila.colorado.edu
* weberjm@jila.colorado.edu