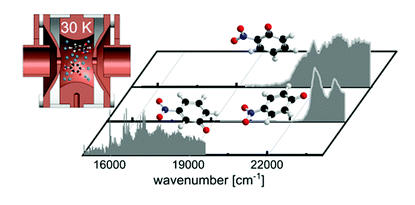
The intrinsic photophysics of nitrophenolate isomers (meta, para, and ortho) was studied at low temperature using photodissociation mass spectrometry in a cryogenic ion trap instrument. Each isomer has distinct photophysics that affects the excited state lifetimes, as observed experimentally in their spectroscopic linewidths. Visible-light-induced excitation of m-nitrophenolate gives rise to well-resolved vibronic features in the spectrum of the S1 state. The para and ortho isomers have broad spectra – even at cryogenic temperatures – due to their shorter excited state lifetimes and spectral congestion. We present computational evidence for mixing of the first and second excited states of o-nitrophenolate, leading to significant additional broadening in the experimental spectrum.
DOI: 10.1039/C8CP06078A
Leah G. Dodson,a Wyatt Zagorec-Marks,b Shuang Xu,c James E. T. Smithb and J. Mathias Weberb
aJILA and NIST, University of Colorado, 0440 UCB, Boulder, USA
bJILA and Department of Chemistry, University of Colorado, 0440 UCB, Boulder, USA
cJILA and Department of Physics, University of Colorado, 0440 UCB, Boulder, USA