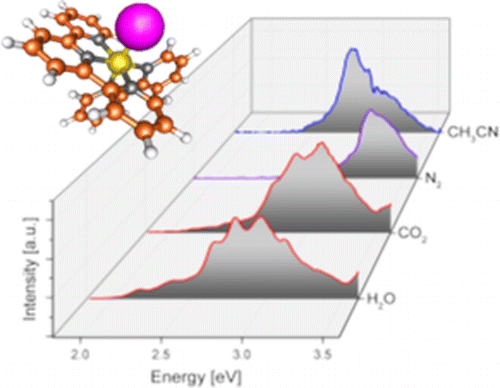
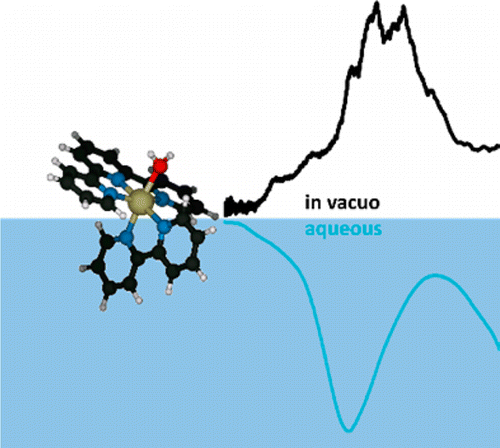
We report the electronic spectra of mass selected [(bpy)(tpy)Ru–OH2]2+·(H2O)n clusters (bpy = 2,2′-bipyridine, tpy =2,2′:6′2″-terpyridine, n = 0–4) in the spectral region of their metal-to-ligand charge transfer bands. The spectra of the mono- and dihydrate clusters exhibit partially resolved individual electronic transitions. The water network forming at the aqua ligand leads to a rapid solvatochromic shift of the peak of the band envelope: addition of only four solvent water molecules can recover 78% of the solvatochromic shift in bulk solution. The sequential shift of the band shows a clear change in behavior with the closing of the first hydration shell. We compare our experimental data to density function theory (DFT) calculations for the ground and excited states.
Shuang Xu†, James E. T. Smith‡, and J. Mathias Weber‡
† JILA and Department of Physics, University of Colorado, Boulder, Colorado 80309-0440, United States
‡ JILA and Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309-0440, United States